
Abstract
介绍分箱是什么,做这篇工作的原因。本文的主要内容是什么,结果怎么样,最终的用途。
Introduction
介绍为什么需要分箱,微生物多样性未知,此外许多很难在实验室隔离培养。所以出现了 binning作为一个策列去探索未知的微生物社区。
介绍了几种binning的方法原理,然后几种整合高质量MAGs的工具,MetaWRAP, DAS Tool MAGScoT。
宏基因组分箱的模式:
co-assembly 联合组装分箱, 将所有样本放到一起组装,然后计算这些contig在每个样本的coverage,
singel-sample 单样本分箱,每个样本单独组装,单独分箱
multi-sample 多样本分箱, 每个样本单独组织,计算所有样本分别的coverage信息。
本文的目的必要性,说了几点之前类似文章的缺陷,没有评估不同数据类型binning模式下不同方法的性能,没有考虑到最新的binning方法和评估方法类似CheckM2
这个工作,评估了13中binning工具,用了7个binning结合方式,在5个真是数据集上测试,数据包括ngs,hifi,和nanopore。
结果证明,mult-sample 比 single-sample 效果提升都很明显。然后是几种binning工具的性能,和bin优化工具的性能。
Results
Benchmarking binners on seven data-binning combinations
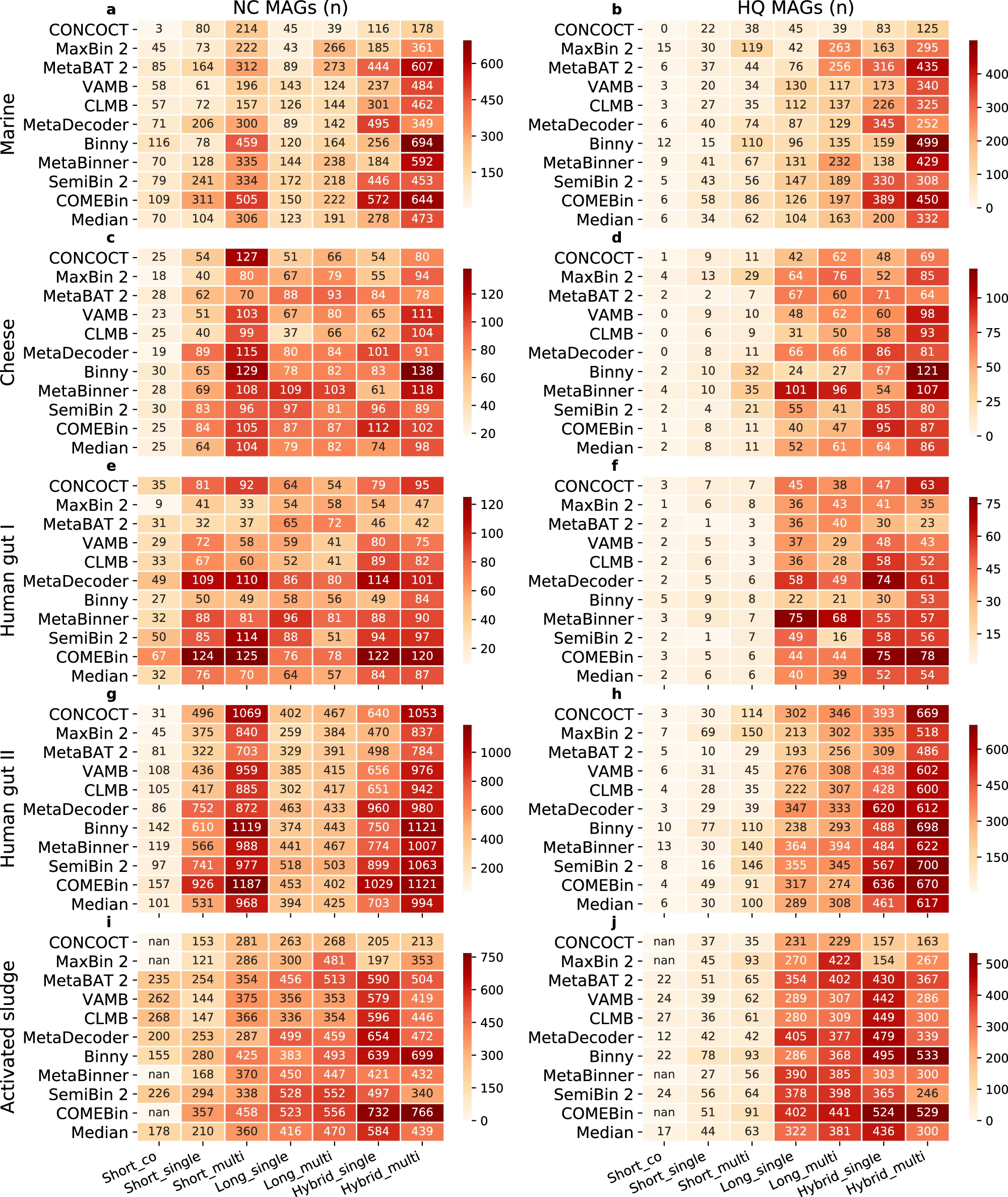
HQ (completeness > 90%, contamination < 5% and presence of the 23S, 16S, and 5S rRNA genes and at least 18 tRNAs),
NC (completeness > 90% and contamination < 5%),
MQ (completeness > 50% and contamination < 10%).
比较了不同数据,不同的分箱模式得到的不同的结果,那种方式结果更好
对于 short read
对于 long read
对于 hybrid data
总结,multi-sample 比 singel-sample 不管是在short read ,long read ,hybrid data上效果都好。另外,short read在HQ MAG上效果一般。不管是single-sample 还是 mult-sample ,hybrid data效果更好,
Determining the high-performance and efficient binners
COMEBin, MetaBinner, and SemiBin 2
说明评估结果,运行时间,总体打分,一些特殊情况,例如几个工具2周没跑完。
哪个工具综合最好,分别数据集上哪个工具最好。
各个工具花费的时间和内存
Identifying the optimal bin-refinement tool
确定最好的 bin-refinement 工具
Multi-sample binning recovers extensive species and strains across diverse data types
Multi-sample binning reveals extensive potential ARG hosts and BGCs across diverse data types
Discussion
Methods
Benchmarking datasets and preprocessing
选择这几个数据集的原因
都有short read 和 long read
来自5个不同的环境
都是公开的
用FastQC和MultiQC检查illumina和hifi read 质量,Nanoplot检查Nanppore的read 质量
对于mNGS样本
Fastp预处理mNGS样本
-q 20 --length_required 100 --low_complexity_filter
对于nanopore样本
qcat 处理裁剪nanopore adapter 和barcode sequence
--trim --detect-middle
裁剪后用Filtlong 去除低质量和短序列
--min_length 4000 and --min_mean_q 80
Porechop 检测nanopore read剩余的adapter和barcodes
--min_split_read_size 4000
最后用Filtlong用前面的参数过滤。
此外,人体内的样本,会去掉能用bowtie2比对到hg38 reference 上的 reads .
Metagenome assembly and alignment
MEGAHIT 进行short read的 co-assembly 和 single-sample组装。
Flye 进行long-read 的 single-sample 。OPERA-MS 用来进行hybrid single-sample 组装。hybrid single-sample 组装意思是每个short read sample和他相关的long-read sample一起组装,hybrid组装结果用,如果用nanopore序列的话,用Pilon抛光。
选出长度大于1kb的contig 进行后续分析。
Bowtie2 比对 short reads, minimap2 比对long reads。
对于 hybrid binning ,short read alignment 和long read alignment 都用做binning 的输入。
samtools 用于sort alignment 文件
Contig binning
介绍了三种 binning模式和binning及其refinement用了哪些工具
Evaluation metrics and ranking score
评估工具,主要用Checkm2评估完整度和污染度
还有一个整体的分数九三公式,结合了各个数据的binning结果
Dereplication and phylogenetic analysis of MAGs
dRep 用来去重
物种级别 species level 95%核苷酸相似性 ANI
-p 32, -nc 0.6 -sa 0.95
菌株水平 strain level 99% 核苷酸相似性 ANI
-p 32, -nc 0.6 -sa 0.99
用GTDB-Tk对MAG进行注释(GTDB r220)。IT-TREE构建进化树,iTOL v7 使用-m LG+R4可视化